Blog
Medical Device Document Control Requirements for FDA and ISO 13485 Compliance

Medical device document control is not just a regulatory checkbox. It is the backbone of your quality system and one of the most common reasons companies face FDA observations, audit delays, and costly compliance gaps. For MedTech leaders, CTOs, and compliance officers, poor document control means risk, inefficiency, and lost time to market.
If your organization is still relying on fragmented systems, manual approvals, or inconsistent version tracking, you are already exposed. FDA document control expectations under 21 CFR 820 and ISO 13485 requirements demand structured, traceable, and audit-ready documentation at every stage of the product lifecycle.
Learn exactly how to implement a compliant and scalable medical device document control system, avoid regulatory pitfalls, and transition from reactive compliance to a proactive, conversion-ready quality strategy that supports growth.
A Closer Look at Document Control in Regulated Environments
Medical device document control is a structured system that governs how documents are created, reviewed, approved, distributed, updated, and archived within a Quality Management System. In regulated environments like MedTech and SaMD documentation is not just operational support. It is legally and clinically critical.
Every document must be accurate, traceable, and accessible during audits or inspections. Even a small inconsistency can trigger compliance issues, delays, or regulatory findings.
This includes:
- SOPs
- Work instructions
- Design and development files
- Regulatory submissions
- Quality and compliance records
A controlled system ensures that only approved and current documents are in use while maintaining a full history of changes.
Difference between document control and document management
Many organizations assume document management tools are enough for compliance. This is a costly misconception. While both concepts are related, they serve very different purposes in regulated industries.
Document management focuses on organization and access, while document control enforces compliance, traceability, and regulatory integrity.
Aspect | Document Control | Document Management |
Purpose | Ensures regulatory compliance and traceability | Organizes and stores documents |
Approval | Mandatory review and approval workflows | Not always required |
Versioning | Strict version control with audit trails | Basic versioning or none |
Compliance | Built for FDA and ISO requirements | Not inherently compliant |
Risk Level | High impact on audits and approvals | Operational convenience |
Why this distinction matters
If your system only manages documents but does not control them:
- You risk using outdated files
- You lack traceability during audits
- You increase the chances of FDA observations
In regulated environments, document management without control is a compliance liability.
Why document control is critical for FDA and ISO compliance
Regulators do not just evaluate what you document. They evaluate how well those documents are controlled throughout their lifecycle.
Both FDA and ISO require organizations to implement structured control mechanisms that ensure consistency, traceability, and accountability.
Key Compliance Requirements
Document approval before use: Every document must be formally reviewed and approved by authorized personnel before it is released into the system. This ensures accuracy and compliance from the start.
Controlled distribution: Only the latest approved versions should be accessible at points of use. This prevents teams from working with outdated or incorrect information.
Change management: Any modification must go through a documented review and approval process. Unauthorized or undocumented changes are a major compliance risk.
Obsolete document control: Outdated documents must be removed from active use but retained for historical and audit purposes.
What happens without proper control?
Organizations often experience:
- Version confusion across departments
- Missing or incomplete audit trails
- Delays in regulatory submissions
- Increased risk of FDA 483 observations
Regulatory Requirements for Document Control FDA and ISO 13485
Under 21 CFR 820.40, the FDA requires medical device manufacturers to establish and maintain strict document control procedures within their quality systems.
These requirements are not optional. They are enforceable and frequently reviewed during inspections.
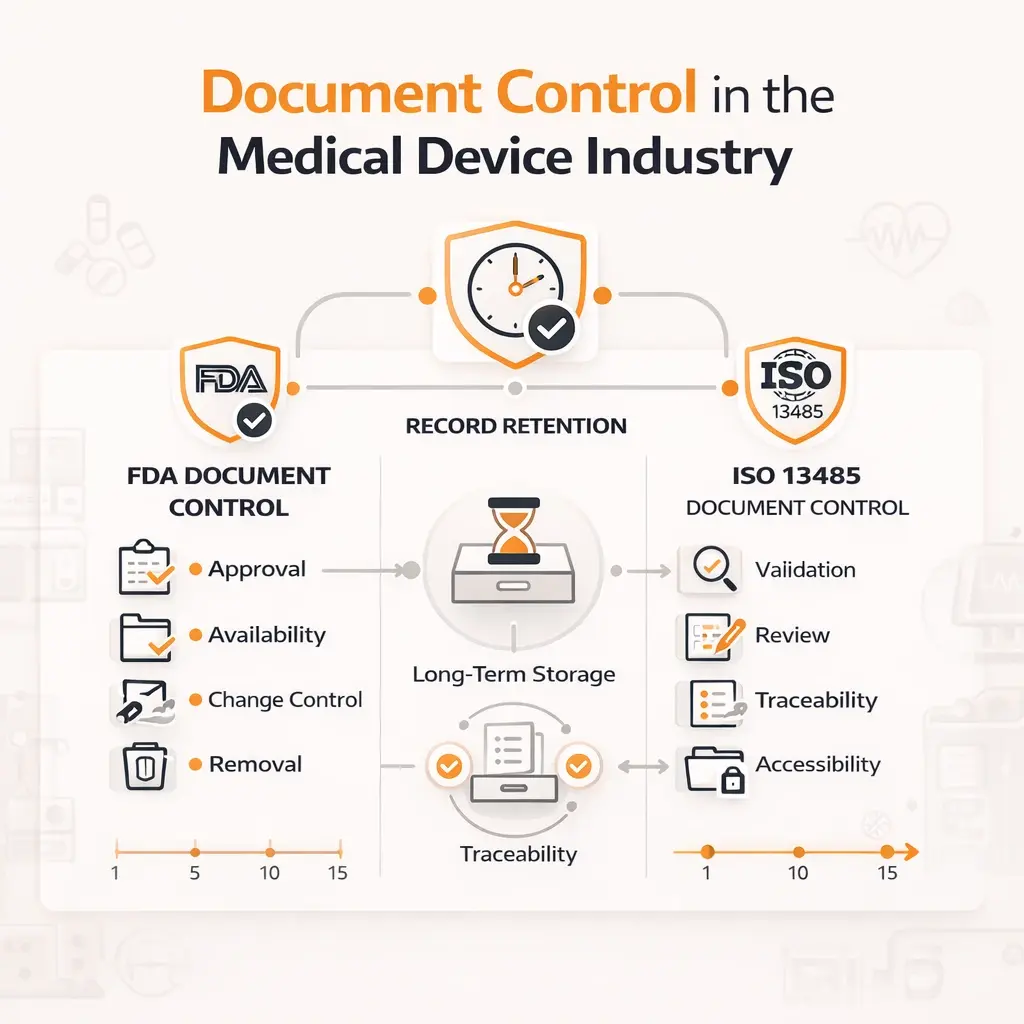
Document approval:
Documents must be reviewed and approved by designated individuals before implementation. Approval must be documented and traceable.
Document availability:
Current versions must be available at all relevant locations where they are used. Teams should never rely on unofficial or outdated copies.
Change control:
All changes must be formally reviewed, approved, and recorded. This includes tracking what changed, who approved it, and when.
Document removal:
Obsolete documents must be promptly removed from operational use to prevent accidental misuse.
1. FDA record retention requirements for medical devices
In addition to controlling documents, the FDA mandates strict retention policies for records related to medical devices.
These requirements ensure long-term traceability and accountability.
Minimum retention period: Records must be retained for at least 2 years from the date of release or for the expected life of the device, whichever is longer.
Applies to critical records: This includes design files, production records, complaint files, and CAPA documentation.
Retrievability requirement: Records must be easily accessible and retrievable during inspections or audits.
2. ISO 13485 document control requirements
ISO 13485 provides a comprehensive framework for document control within a medical device QMS. It emphasizes consistency, traceability, and continuous improvement.
Documents must be validated for accuracy and compliance before being issued. Also, documents should be regularly reviewed to ensure they remain current and relevant. All modifications must be clearly documented and traceable. Relevant documents must be accessible where they are needed operationally. Documents must remain readable and properly labeled throughout their lifecycle.
3. Key differences and overlaps between FDA and ISO
Both FDA and ISO share a common goal: ensuring controlled, reliable documentation. However, their approach and emphasis differ slightly.
FDA requirements are enforcement-driven and inspection-focused, while ISO 13485 is framework-driven and system-focused. Together, they create a comprehensive compliance landscape that organizations must navigate.
Criteria | FDA 21 CFR 820.40 | ISO 13485 |
Approach | Regulatory enforcement | Quality management framework |
Focus | Inspection readiness | System consistency |
Flexibility | Prescriptive | Flexible implementation |
Audit Style | FDA inspections | Certification audits |
Objective | Legal compliance | Continuous improvement |
Types of Documents in the Medical Device Industry
There are three document types that form the backbone of medical device traceability and compliance. Together, they provide a complete record of design, production, and manufacturing outcomes.
- I) Design History File (DHF):
Contains all records demonstrating that the device was designed according to regulatory and design control requirements. - II) Device Master Record (DMR):
Defines how the device should be manufactured, including specifications, processes, and quality procedures.
III) Device History Record (DHR):
Provides evidence that each manufactured device meets the specifications outlined in the DMR.
Corrective and Preventive Action CAPA records
CAPA records are essential for identifying, correcting, and preventing quality issues within a medical device organization.
They must be tightly controlled to ensure reliability and compliance.
- Issue identification: Capturing problems from complaints, audits, or internal findings.
- Root cause analysis: Determining the underlying reason behind the issue.
- Corrective action: Implementing fixes to resolve the issue.
- Preventive action: Ensuring the issue does not recur in the future.
For a deeper understanding, see Corrective and Preventive Action (CAPA) in medical devices. Without proper document control, CAPA systems lose effectiveness and traceability.
SOPs Work Instructions & Quality Records
Operational documentation ensures consistency and repeatability across processes.
- Standard Operating Procedures (SOPs): Define high-level processes and policies within the organization.
- Work instructions: Provide detailed, step-by-step guidance for executing tasks.
- Quality records: Capture evidence that processes were performed as required.
Version control and audit trails
Version control and audit trails are essential components of an effective medical device document control system, as they ensure full transparency and traceability across all documentation activities.
Version control guarantees that only the most recent, approved version of a document is actively used within the organization, while securely maintaining access to previous versions for reference and audit purposes. At the same time, audit trails provide a complete, time-stamped record of all document-related actions, including changes, approvals, and user access.
Together, these mechanisms not only support regulatory compliance with FDA and ISO 13485 requirements but also strengthen accountability, reduce errors, and ensure the organization remains fully audit-ready at all times
Medical Device Document Control Process Step by Step
A compliant medical device document control process is not just about storing files. It is a structured lifecycle that ensures every document is accurate, approved, traceable, and audit-ready at all times.
For MedTech and SaMD companies, this process must align with both FDA expectations and ISO 13485 requirements while remaining scalable as the organization grows.
Below is a practical, real-world workflow used by high-performing, audit-ready organizations.
1. Document creation and standardization
The process begins with structured document creation. Without standardization at this stage, everything downstream becomes inconsistent and difficult to control.
- Standardized templates:
Use predefined formats for SOPs, work instructions, and records to ensure consistency across all departments and teams. - Defined ownership:
Each document must have a clearly assigned owner responsible for accuracy, updates, and compliance. - Controlled document identification:
Unique IDs, naming conventions, and version labels must be assigned to avoid duplication and confusion.
2. Review and approval workflows
Once a document is created, it must go through a formal review and approval process before it can be used.
- Multi-level review:
Subject matter experts, QA, and compliance teams validate accuracy, completeness, and regulatory alignment. - Approval authorization:
Only designated personnel can approve documents, ensuring accountability and control. - Electronic signatures and tracking:
Approvals must be recorded with timestamps and user identification for audit traceability.
3. Distribution and access control
After approval, documents must be distributed in a controlled manner to ensure only the correct versions are used.
- Role-based access:
Employees should only access documents relevant to their responsibilities, reducing misuse and confusion. - Centralized repository:
All documents must be stored in a single, controlled system to eliminate duplicates and inconsistencies. - Real-time updates:
When a document is revised, users should automatically access the latest version without manual intervention.
4. Version control and change management
Every document will evolve over time. The key is controlling how those changes are introduced and tracked.
- Version tracking:
Each update must generate a new version with clear documentation of changes made. - Change justification:
Every revision should include a reason for the change, linking it to audits, CAPA, or process improvements. - Approval of revisions:
Changes must go through the same approval workflow as new documents.
5. Document archival and retention
The final stage ensures that documents are properly stored, retained, and retrievable for audits and inspections.
- Secure archival systems:
Obsolete documents must be stored in a way that prevents accidental use but allows retrieval when needed. - Retention policies:
Documents must be retained according to FDA and ISO requirements, including device lifecycle considerations. - Audit-ready retrieval:
Documents should be easily searchable and accessible during inspections without delays.
Common Document Control Challenges in MedTech Companies
Even with documented procedures in place, many MedTech and SaMD companies struggle to maintain effective document control as they scale. What looks compliant on paper often breaks down in execution, especially when teams grow, products evolve, and regulatory pressure increases.
The reality is that most document control failures are not caused by lack of intent. They are caused by inefficient systems, manual processes, and disconnected workflows that cannot keep up with compliance demands.
Manual processes and version confusion
Many organizations still rely on shared drives, email approvals, and static file storage systems to manage critical documentation. While this may work in early stages, it quickly becomes unmanageable as complexity increases.
Manual approvals via email: Documents are reviewed and approved through email threads, making it difficult to track decisions, maintain records, and ensure accountability.
Multiple document versions: Teams often store files locally or across different folders, leading to multiple conflicting versions of the same document.
Lack of real-time updates: Employees may unknowingly use outdated documents because there is no centralized system enforcing the latest version.
Audit failures and FDA 483 risks
Document control is one of the first areas reviewed during FDA inspections and ISO audits. Weaknesses here often result in immediate findings.
Missing approval records: Inability to demonstrate that documents were properly reviewed and approved before use.
Incomplete audit trails: Lack of visibility into who made changes, when they were made, and what was modified.
Uncontrolled obsolete documents: Outdated documents still accessible or in use within the organization.
Lack of traceability across systems: As organizations adopt multiple tools for quality, regulatory, and operational processes, documentation often becomes fragmented.
Disconnected systems: Document control, CAPA, risk management, and design controls are managed in separate platforms without integration.
Broken traceability links: Difficulty connecting documents to specific processes, changes, or regulatory requirements.
Data silos: Critical information is scattered across teams, tools, and departments.
Scaling issues in growing organizations
What works for a small team often fails at scale. As MedTech companies grow, document control must evolve to handle increased complexity, volume, and regulatory exposure.
Increased document volume: More products, processes, and teams generate a higher number of documents to manage.
Cross-functional collaboration: Multiple departments need access to controlled documents, increasing the risk of inconsistencies.
Global compliance requirements: Organizations operating across regions must align with both FDA and ISO standards simultaneously.
How Document Control Software eQMS Solves These Challenges
As MedTech companies scale and regulatory expectations increase, manual and fragmented document control systems quickly become unsustainable. What starts as a manageable process often turns into a bottleneck that slows approvals, increases compliance risk, and limits organizational growth.

This is where document control software, commonly part of an electronic Quality Management System (eQMS), becomes essential. Instead of relying on disconnected tools and manual oversight, an eQMS embeds compliance directly into your workflows.
What is a document control system software?
Document control system software is a centralized platform designed to manage the entire lifecycle of controlled documents within a regulated environment. Unlike basic storage tools, it enforces compliance through automation, traceability, and structured workflows.
Features of a regulatory document management system
A regulatory-grade document control system goes beyond basic functionality. It is specifically designed to meet FDA and ISO requirements while supporting complex MedTech workflows.
Role-based access control:
Ensures users only access documents relevant to their roles, improving security and reducing misuse.
Electronic signatures and approval logs:
Captures legally compliant approvals with timestamps and user identification for audit readiness.
Integrated change control:
Links document updates to CAPA, audits, and process improvements for complete traceability.
Automated notifications:
Alerts users about pending reviews, approvals, or updates to prevent delays and bottlenecks.
Compliance alignment:
Built-in support for standards like 21 CFR 820 and ISO 13485 ensures regulatory requirements are met by design.
Benefits of healthcare document management software
Implementing a modern document control system delivers measurable benefits across compliance, operations, and business performance.
- Improved compliance posture
- Faster approval cycles
- Enhanced traceability
- Reduced operational risk
- Scalability
How eQMS ensures compliance and scalability
An eQMS does not just digitize document control. It standardizes and enforces it across the entire organization.
It connects document control with other critical quality processes, creating a unified compliance ecosystem.
- CAPA systems:
Automatically link document updates to corrective and preventive actions to maintain alignment. - Design controls:
Ensure documents related to DHF, DMR, and DHR remain consistent and traceable throughout the product lifecycle. - Risk management:
Align documentation with risk assessments and mitigation strategies. - Software lifecycle processes:
For SaMD organizations, integration with standards like IEC 62304 software lifecycle requirements ensures software documentation remains compliant.
Best Practices for Implementing Document Control in Medical Devices
Implementing document control is not just about meeting regulatory requirements, it’s about building a system that is consistent, scalable, and audit-ready by design. Organizations that pass audits smoothly typically have well-structured, integrated processes, while those that struggle often lack alignment and standardization. Strong document control creates clarity, reduces risk, and ensures long-term compliance.
Aligning with ISO 13485 and FDA Expectations
A robust document control system must satisfy both FDA and ISO requirements simultaneously. Treating these frameworks separately often leads to inefficiencies, duplication, and compliance gaps.
By unifying processes, standardizing documentation structures, and clearly defining roles and responsibilities, organizations can build a cohesive system that ensures accountability and consistency.
This approach reduces redundant work, minimizes regulatory risks, and simplifies audit preparation—making compliance more efficient and reliable.
Integrating CAPA, Risk Management, and Design Controls
Document control should not function in isolation; it must be deeply integrated with key quality processes. Linking document updates with CAPA ensures corrective actions are properly documented and traceable.
Integrating risk management ensures that documentation reflects current risk assessments and mitigation strategies. Aligning with design controls ensures consistency across DHF, DMR, and DHR throughout the product lifecycle.
Without this integration, systems become fragmented and traceability weakens. When properly connected, document control becomes a single source of truth across the entire QMS.
Preparing for Audits and Inspections
Audit readiness should be embedded into the system from the start, not treated as a periodic task. A strong document control system maintains complete audit trails, ensures instant document retrieval, and properly manages obsolete documents to prevent misuse.
Auditors typically look for clear approval workflows, version histories, and consistency across documentation. If these elements cannot be demonstrated immediately, organizations risk non-compliance. Building audit readiness into daily operations ensures confidence during inspections.
Continuous Improvement and Quality Assurance
Document control is an evolving process that must adapt to changes in products, regulations, and organizational growth. Regular document reviews help maintain accuracy and relevance, while performance monitoring highlights inefficiencies and improvement opportunities.
Incorporating feedback from teams ensures processes remain practical and effective. Over time, continuous improvement strengthens compliance, enhances efficiency, and reduces regulatory risk, transforming document control from a burden into a strategic advantage.
Choosing the Right Document Control System for Your Organization
Selecting the right document control system is a critical decision that directly impacts compliance, efficiency, and scalability. The wrong choice can create long-term limitations, while the right system can accelerate growth and reduce regulatory risk.
Key features to look for:
Not all systems are built for regulated industries. MedTech companies need solutions specifically designed for compliance.
Regulatory compliance support: The system should align with FDA 21 CFR 820 and ISO 13485 requirements out of the box.
Workflow automation: Automate document review, approval, and change processes to reduce manual effort.
Version control and audit trails: Ensure complete traceability for every document and action.
Scalable architecture: The system should support growing teams, increasing document volume, and global operations.
Build vs Buy Custom vs Off the Shelf Solutions
One of the biggest decisions organizations face is whether to build a custom system or adopt an existing solution.
Off-the-shelf systems offer speed and pre-built compliance features, while custom solutions provide flexibility and alignment with specific business needs.
Criteria | Off-the-Shelf eQMS | Custom-Built Solution |
Deployment Speed | Fast implementation | Longer development time |
Compliance Readiness | Pre-configured for FDA/ISO | Requires custom validation |
Flexibility | Limited customization | Fully tailored to needs |
Cost | Lower upfront | Higher initial investment |
Scalability | Depends on vendor | Fully scalable if designed well |
Summary
Medical device document control is often viewed as a regulatory obligation. In reality, it is a strategic function that directly impacts your organization’s ability to scale, innovate, and compete.
Companies that treat document control as a checkbox struggle with audits, delays, and inefficiencies. Those who invest in structured, integrated, and scalable systems gain a clear advantage.
They move faster. They operate with confidence. They pass audits without disruption.
Table of Contents
1) A Closer Look at Document Control in Regulated Environments
2) Regulatory Requirements for Document Control FDA and ISO 13485
3) Types of Documents in the Medical Device Industry
4) Medical Device Document Control Process Step by Step
5) Common Document Control Challenges in MedTech Companies
6) How Document Control Software eQMS Solves These Challenges
7) Best Practices for Implementing Document Control in Medical Devices
8) Choosing the Right Document Control System for Your Organization
9) Summary
Innovate the Future of Health Tech
CitrusBits helps MedTech leaders build smarter apps, connected devices, and XR health solutions that truly make an impact.
