Blog
CAPA Medical Device Best Practices for Compliance and Audits

Corrective and Preventive Action (CAPA) is a core regulatory requirement for any medical device company, and it becomes even more critical when dealing with Software as a Medical Device. From software bugs that impact clinical performance to post-market surveillance signals flagged by users or regulators, CAPA is how organizations identify issues, correct them, and prevent recurrence.
For MedTech companies operating across FDA and ISO-regulated markets, CAPA is not just a quality checkbox. It is a foundational system that demonstrates control, accountability, and patient safety.
Regulators expect CAPA processes to be proactive, data-driven, and fully integrated into your quality management system, especially when software updates, validation cycles, and cybersecurity risks are involved.
What Is CAPA in Medical Devices?
In the medical device industry, CAPA refers to a formal, documented process used to investigate quality issues, identify root causes, implement corrective actions, and establish preventive measures to stop problems from happening again. CAPA is required under FDA Quality System Regulations and ISO 13485, making it a non-negotiable component of compliance.
CAPA often extends beyond physical defects and includes:
- Software failures affecting clinical logic
- Validation gaps introduced by updates
- Post-market complaints tied to usability or performance
- Cybersecurity vulnerabilities with patient safety impact
A well-functioning CAPA process demonstrates that an organization can detect issues early, analyze them effectively, and maintain ongoing control over its product throughout the software lifecycle.
What Does CAPA Stand For?
CAPA stands for Corrective and Preventive Action.
- Corrective Action addresses an existing problem by eliminating its root cause.
- Preventive Action focuses on identifying potential issues and stopping them before they occur.
Regulators expect both elements to work together. Fixing a problem without preventing recurrence is considered an incomplete CAPA.
What Is a CAPA in Quality Management Systems?
Within a Quality Management System (QMS), a CAPA is a controlled quality event that follows a defined workflow. This typically includes:
- Issue identification (complaints, audits, nonconformances)
- Root cause analysis
- Action planning and implementation
- Effectiveness verification
- Documentation and closure
CAPA records are among the first things auditors review because they reveal how well an organization responds to risk, change, and failure.
For MedTech teams, CAPA must integrate with:
- Change control
- Software validation
- Risk management
- Post-market surveillance
This ensures that software updates and corrective fixes do not introduce new compliance or safety risks.
What Does CAPA Mean in Healthcare and SaMD?
In healthcare and SaMD contexts, CAPA directly supports patient safety and clinical reliability. Unlike traditional hardware-focused devices, SaMD products evolve rapidly, making continuous monitoring and improvement essential.
- Algorithm errors impacting clinical decisions
- Inadequate validation after software updates
- User-reported issues from real-world use
- Regulatory findings from FDA inspections or ISO audits
Because software changes can be deployed quickly, regulators expect SaMD companies to have tight CAPA controls that balance speed with compliance. A weak CAPA process is often interpreted as a broader failure in quality governance.
Why CAPA Is Critical for SaMD Companies
For Software as a Medical Device (SaMD) companies, CAPA is not just a regulatory formality. It is a critical control mechanism that ensures software performance, clinical safety, and ongoing compliance across the product lifecycle.
Unlike traditional medical devices, SaMD products evolve rapidly. Frequent updates, algorithm changes, integrations, and cybersecurity patches increase the likelihood of introducing new risks. CAPA provides a structured way to manage these risks while maintaining regulatory confidence.
For companies developing Medical Devices, regulators expect CAPA to be tightly connected to software development, validation, and post-market activities. A weak or reactive CAPA process is often interpreted as a sign that the organization lacks control over its software lifecycle.
Regulatory Expectations for CAPA in Software-Based Medical Devices
Regulators evaluate CAPA as evidence that a SaMD company can:
- Detect quality and safety issues early
- Investigate issues beyond surface-level symptoms
- Implement changes without introducing new risks
- Verify that corrective actions actually work
In SaMD environments, CAPA commonly intersects with:
- Software defect tracking and resolution
- Risk management updates
- Change control and revalidation
- Post-market surveillance and complaint handling
FDA inspectors frequently link CAPA deficiencies to broader systemic failures, such as inadequate software validation or ineffective risk management. This makes CAPA one of the highest-risk areas during inspections.
When Should a CAPA Be Initiated?
A CAPA should be initiated whenever there is evidence of a systemic or recurring issue, or when a problem has the potential to impact patient safety or regulatory compliance.
- Repeated software defects affecting functionality
- Validation failures following updates or patches
- Customer complaints indicating usability or performance issues
- Audit findings or regulatory observations
- Post-market data showing unexpected clinical behavior
Not every issue requires a CAPA. However, failing to initiate one when warranted is a common reason for FDA 483 observations. Regulators expect organizations to apply risk-based judgment and clearly justify when CAPA is or is not opened.
FDA and ISO Requirements for CAPA
CAPA is a mandatory requirement under both FDA Quality System Regulations and ISO 13485. While the language differs slightly, the intent is the same: manufacturers must have a documented system to identify problems, correct them, and prevent recurrence.
For companies operating in multiple regions, CAPA must be designed to satisfy both FDA and ISO expectations simultaneously, especially when SaMD products are marketed globally.
FDA CAPA Requirements
Under FDA regulations, CAPA is governed by 21 CFR 820.100. The FDA expects manufacturers to:
- Analyze quality data to identify existing and potential problems
- Investigate root causes using appropriate methodologies
- Implement corrective and preventive actions
- Verify or validate actions to ensure effectiveness
- Document activities and maintain records
FDA scrutiny often focuses on:
- How software issues are detected and escalated
- Whether corrective actions are properly validated
- How changes are controlled and documented
- Whether CAPA outcomes feed back into risk management
CAPA is one of the most cited areas in FDA warning letters, particularly when organizations close CAPAs without verifying effectiveness.
What Is CAPA in ISO 13485?
ISO 13485 requires organizations to establish a CAPA process under Clause 8.5. The standard emphasizes:
- Systematic investigation of nonconformities
- Determination of root causes
- Implementation of actions proportional to risk
- Review of CAPA effectiveness
For SaMD companies, ISO auditors expect CAPA to align with:
- Software lifecycle processes
- Risk management under ISO 14971
- Change management and validation controls
ISO 13485 places strong emphasis on documentation and consistency. CAPA records must clearly show decision-making, risk evaluation, and follow-through.
Multi-Region CAPA Compliance (FDA + ISO Alignment)
For global SaMD companies, the most effective approach is to implement a single CAPA system that satisfies both FDA and ISO requirements.
Best practices include:
- Using a unified CAPA workflow mapped to both regulations
- Aligning CAPA severity with risk management outputs
- Ensuring audit readiness across regions
- Maintaining traceability between CAPA, change control, and validation
A harmonized CAPA system reduces duplication, lowers audit risk, and supports scalable growth across regulated markets.
The CAPA Process Explained
The CAPA process is a structured, closed-loop system used by medical device organizations to identify quality issues, correct them at the root level, and prevent recurrence. Regulators evaluate not only whether CAPAs are opened, but whether the CAPA process in medical devices is effective, risk-based, and consistently applied across the organization.
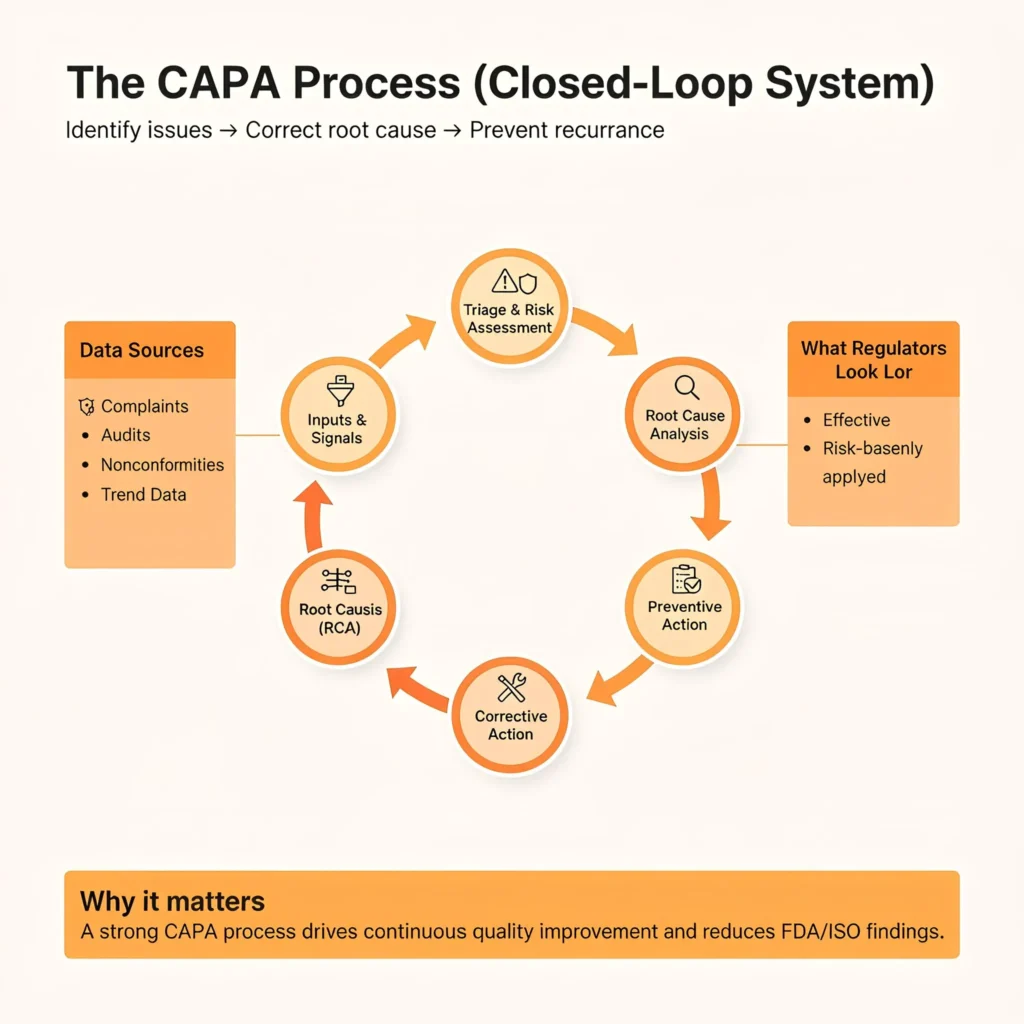
A well-designed CAPA process supports continuous quality improvement by linking complaints, audits, nonconformities, and trend data into actionable corrective and preventive actions. In contrast, an informal or poorly documented CAPA process is one of the most common reasons for FDA inspection findings and ISO 13485 nonconformities.
From a regulatory perspective, CAPA process improvement is evidence that a manufacturer understands its risks and maintains control over its quality management system
What Are the Four Stages of CAPA?
Although terminology may vary between organizations, the CAPA process for medical devices typically follows four core stages. These stages form a logical CAPA process flow chart that regulators expect to see reflected in documentation and records.
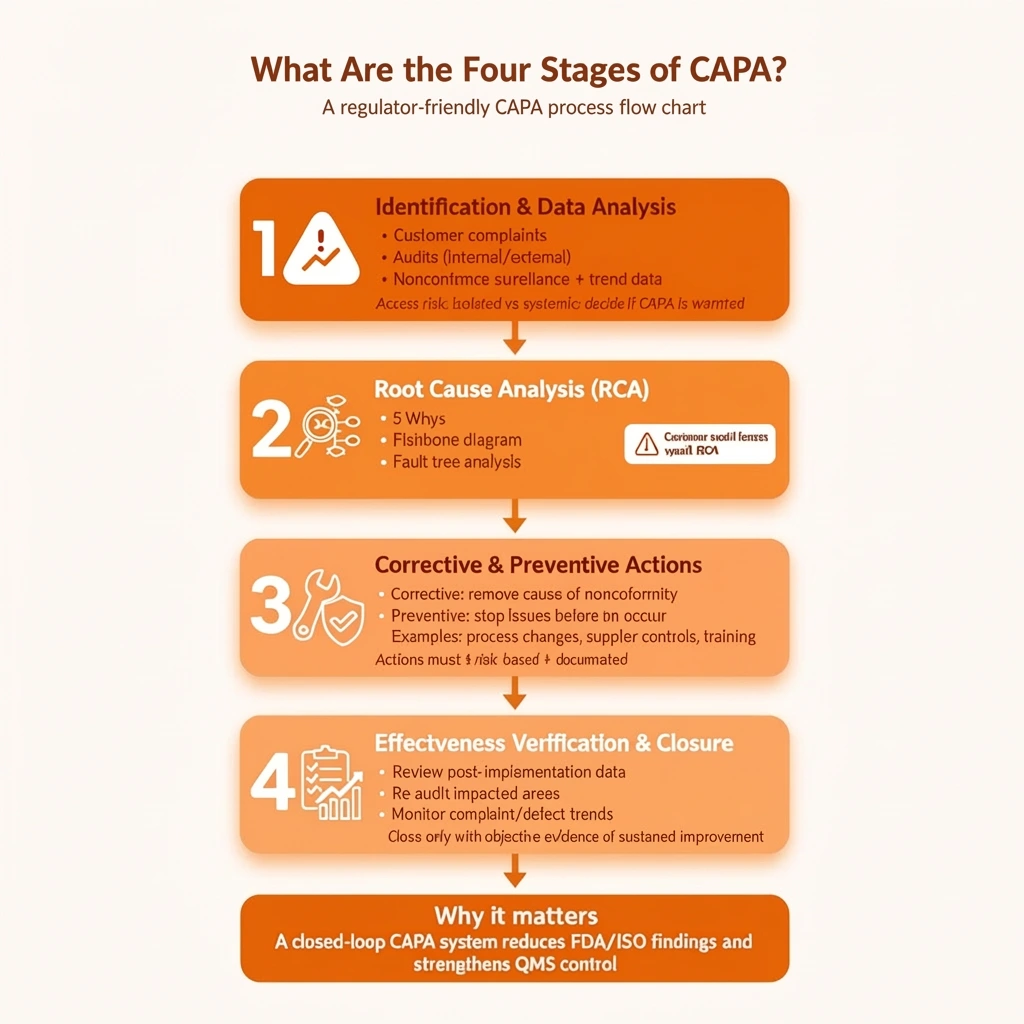
1. Identification and Data Analysis
The CAPA process begins with identifying existing or potential quality issues. These may be detected through:
- Customer complaints
- Internal or external audits
- Nonconformance reports
- Post-market surveillance
- Trend analysis of quality data
At this stage, organizations must evaluate whether the issue is isolated or systemic and whether CAPA initiation is warranted based on risk.
2. Root Cause Analysis (RCA)
Once a CAPA is initiated, the organization must determine the true root cause of the problem. Regulators expect root cause analysis to go beyond symptoms and address underlying process, design, or system failures.
Common RCA methods include:
- 5 Whys
- Fishbone diagrams
- Fault tree analysis
Inadequate root cause analysis is one of the most frequently cited CAPA deficiencies during inspections.
3. Corrective and Preventive Actions
After identifying the root cause, corrective and preventive actions are defined and implemented.
- Corrective actions eliminate the cause of an identified nonconformity.
- Preventive actions address potential issues before they occur.
Actions may include process changes, design updates, supplier controls, training, or revised procedures. All actions must be proportionate to risk and properly documented.
4. Effectiveness Verification and Closure
Before a CAPA can be closed, organizations must verify that the actions taken were effective and did not introduce new risks. Effectiveness verification may involve:
- Reviewing post-implementation data
- Re-auditing affected processes
- Monitoring complaint or defect trends
CAPAs should remain open until objective evidence confirms sustained improvement.
CAPA Process Flow Chart
CAPA Stage | Purpose | Key Outputs |
Identification & Analysis | Detect and assess quality issues | CAPA initiation record, risk evaluation |
Root Cause Analysis | Identify underlying causes | RCA documentation |
Corrective & Preventive Actions | Eliminate causes and prevent recurrence | Action plans, implementation records |
Effectiveness & Closure | Confirm sustained improvement | Verification evidence, CAPA closure |
Why CAPA Process Improvement Matters
Effective CAPA process improvement demonstrates maturity, compliance readiness, and ongoing control over quality risks. Regulators view CAPA as a direct reflection of how well a medical device organization understands and manages its operations.
A consistent, well-documented CAPA process reduces audit risk, supports regulatory approvals, and protects patient safety.
Are CAPA and Root Cause Analysis (RCA) the Same?
CAPA and RCA are closely related but not the same.
- Root Cause Analysis is a method used within the CAPA process to identify why an issue occurred.
- CAPA is the broader system that includes investigation, action planning, implementation, and effectiveness checks.
A common audit finding is treating RCA as the entire CAPA. Regulators expect RCA to feed into meaningful corrective and preventive actions, not replace them.
CAPA Systems and CAPA Management Best Practices
A CAPA system provides the structure and controls needed to manage CAPA activities consistently. For SaMD companies, this system must integrate with software development, quality management, and regulatory reporting.
An effective CAPA management approach demonstrates control, accountability, and continuous improvement.
What Is a CAPA System in Medical Devices?
A CAPA system is the formal framework used to manage CAPA activities from initiation through closure. It typically includes:
- Defined workflows and responsibilities
- Documentation and record control
- Integration with complaints, audits, and risk management
- Effectiveness tracking
CAPA Management for SaMD Teams
Managing CAPA in SaMD environments requires cross-functional collaboration. Quality, engineering, product, and regulatory teams must work together to ensure that actions are effective and compliant.
Best practices for SaMD CAPA management include:
- Clear criteria for when CAPA is required
- Risk-based prioritization of issues
- Formal review and approval processes
- Verification that software changes meet validation requirements
Many SaMD organizations use medical device QMS software to centralize CAPA management, reduce manual errors, and maintain audit readiness.
Is CAPA Part of Six Sigma?
CAPA is not exclusive to Six Sigma, but the concepts are complementary.
- CAPA is a regulatory requirement in medical device quality systems.
- Six Sigma provides tools and methodologies that can support root cause analysis and process improvement.
Some organizations use Six Sigma techniques within their CAPA process, but regulators evaluate CAPA independently of whether Six Sigma is formally adopted.
CAPA Examples in Medical Devices
In medical device organizations, CAPA may be initiated in response to manufacturing issues, design nonconformities, supplier problems, complaints, or post-market surveillance data. Regardless of device type, regulatory expectations for CAPA remain consistent.
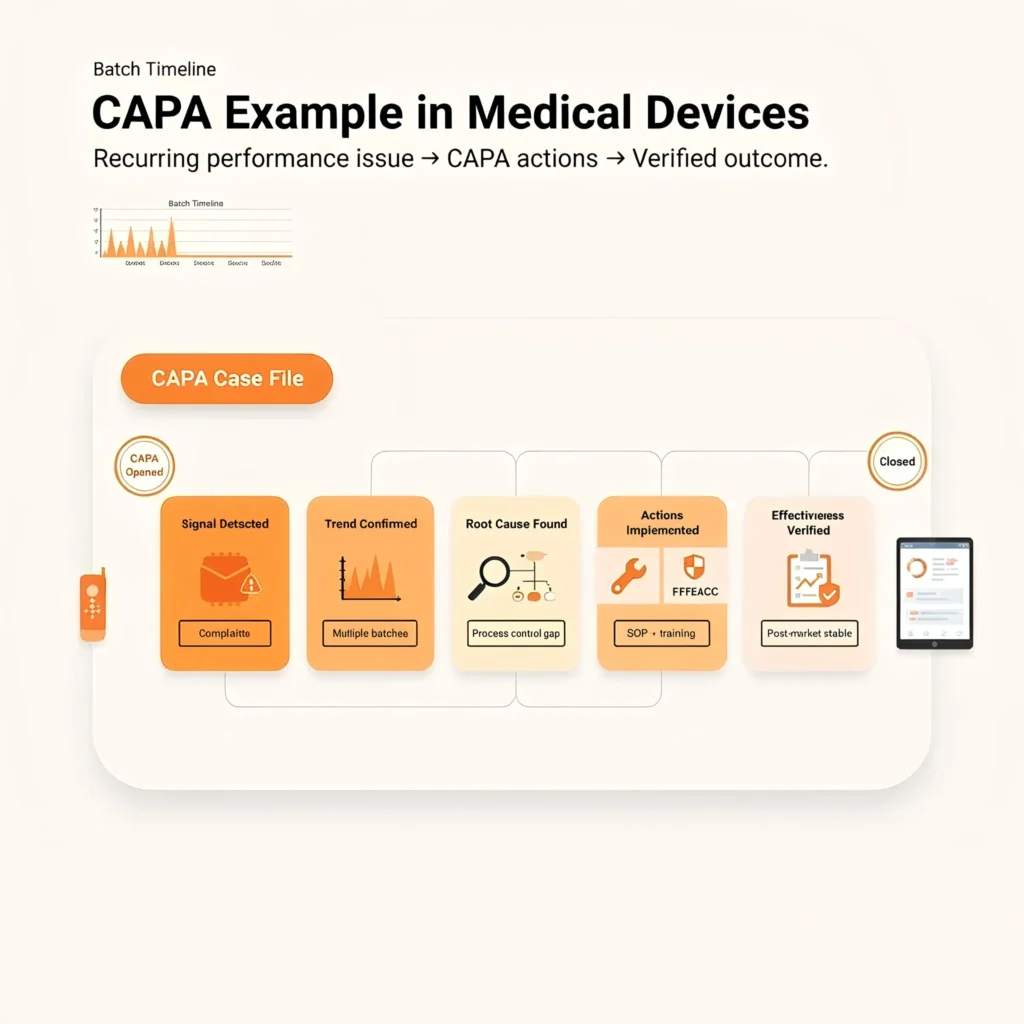
CAPA Example in Medical Devices
Consider a medical device manufacturer that identifies a recurring issue through customer complaints related to inconsistent device performance under specific use conditions. Trend analysis shows the issue occurs across multiple production batches.
A CAPA in this scenario may include:
- Investigation of device design, manufacturing processes, or testing methods
- Root cause identification tied to inadequate process controls or validation gaps
- Corrective action involving design updates, process changes, or revised testing procedures
- Preventive action such as updating standard operating procedures or training programs
- Effectiveness checks confirming the issue no longer occurs in post-market data
This type of CAPA demonstrates regulatory control over product quality, risk management, and patient safety.
CAPA Audit Readiness and Documentation
Auditors expect CAPA documentation to clearly demonstrate:
- Why the CAPA was initiated
- How the root cause was identified
- What corrective and preventive actions were taken and why
- Evidence that actions were verified or validated for effectiveness
- A clear and justified rationale for CAPA closure
Well-documented CAPA records are critical during FDA inspections and ISO 13485 audits, as they show how an organization identifies risk, responds to nonconformities, and maintains control over its quality system.
Common CAPA Mistakes That Lead to FDA Findings
CAPA is one of the most frequently cited problem areas during FDA inspections. Many findings stem from process weaknesses rather than isolated errors.
Common CAPA Compliance Failures
Regulators frequently identify the following issues:
- Superficial root cause analysis that does not address systemic issues
- Closing CAPAs without verifying effectiveness
- Inadequate documentation or missing records
- Failure to initiate CAPA when trends indicate recurring problems
- Poor integration between CAPA, risk management, and change control
In SaMD environments, these failures often coincide with software validation gaps or uncontrolled updates.
How to Avoid CAPA Audit Issues?
To reduce audit risk, SaMD companies should:
- Apply consistent criteria for CAPA initiation
- Use structured root cause analysis methods
- Ensure corrective actions are validated
- Verify effectiveness before closure
- Maintain clear traceability across quality systems
A proactive CAPA approach signals maturity and compliance readiness to regulators.
What Does CAPA Certified Mean?
The term CAPA certified is commonly used in the medical device industry, but it can be misleading. There is no official CAPA certification issued by the FDA, ISO, or any regulatory authority. Instead, CAPA is a mandatory quality system process that is evaluated as part of overall regulatory compliance.
When organizations say they are “CAPA certified,” they usually mean that their CAPA process has been reviewed and found compliant during:
- An FDA inspection under 21 CFR 820
- An ISO 13485 audit conducted by a notified or certification body
In both cases, regulators assess whether the CAPA medical device process is effective, risk-based, and properly documented.
CAPA Certification in Medical Devices: What It Really Means
Rather than a standalone certification, CAPA is reviewed as part of a company’s Quality Management System (QMS). Auditors evaluate whether:
- CAPAs are initiated when required
- Root causes are properly identified
- Corrective and preventive actions are implemented
- Effectiveness is verified before closure
- CAPA records demonstrate control and traceability
A compliant CAPA system strengthens audit outcomes, while CAPA deficiencies are one of the most common causes of FDA 483 observations and ISO 13485 nonconformities
CAPA Certification vs CPAN Certification
Some confusion arises between CAPA certification and other professional or industry credentials. The table below clarifies the difference.
Aspect | CAPA Certification | CPAN Certification |
Issued by | No official issuing body | Certification organizations |
Regulatory status | Not a formal certification | Professional credential |
Scope | CAPA process within a QMS | Individual knowledge or role-based certification |
Evaluated by | FDA inspectors or ISO auditors | Training or certification bodies |
Applies to | Medical device organizations | Individuals, not quality systems |
Purpose | Demonstrates compliance during audits | Demonstrates personal expertise |
Final Thoughts
CAPA is a cornerstone of medical device compliance and a critical safeguard for patient safety, especially in Software as a Medical Device environments where change is constant. A well-designed CAPA system enables organizations to identify risks early, respond effectively, and demonstrate ongoing control to regulators.
For SaMD companies operating under FDA and ISO requirements, CAPA should be integrated into development, validation, and post-market processes rather than treated as a reactive task.
If your organization is building or scaling SaMD products and needs support aligning quality systems with regulatory expectations, explore how CitrusBits helps digital health companies design compliant, scalable medical device solutions.
Table of Contents
1) What Is CAPA in Medical Devices?
2) Why CAPA Is Critical for SaMD Companies
3) FDA and ISO Requirements for CAPA
4) The CAPA Process Explained
5) CAPA Systems and CAPA Management Best Practices
6) CAPA Examples in Medical Devices
7) Common CAPA Mistakes That Lead to FDA Findings
8) What Does CAPA Certified Mean?
9) Final Thoughts
Innovate the Future of Health Tech
CitrusBits helps MedTech leaders build smarter apps, connected devices, and XR health solutions that truly make an impact.
